
期刊:Nature Communications
影响因子:16.6
伯豪技术服务:mRNA检测
导读
胃癌(GC)是世界上最常见的恶性肿瘤之一,其转移速度快,死亡率高。迫切需要GC的诊断标志物和潜在的治疗靶点。究发现ACTL6A对于调节谷胱甘肽 (GSH) 代谢途径至关重要,它上调γ-谷氨酰半胱氨酸连接酶催化亚基 (GCLC) 的表达,从而降低活性氧 (ROS) 水平并抑制铁死亡。机制研究表明,ACTL6A作为核因子(红细胞衍生2)样2 (NRF2)的共转录因子上调GCLC,且ACTL6A的疏水区起重要作用。本研究数据强调了 ACTL6A 在 GC 中的致癌作用,并表明抑制 ACTL6A 或 GCLC 可能是 GC 的潜在治疗策略。
研究技术
mRNA microarray、LC-MS、CHIP-seq
研究设计图
研究结果
1. ACTL6A在胃癌中高表达并促进胃癌进展
从基因表达总集(GEO)中的GC数据集GSE13911、GSE27342、GSE13861获得的mRNA表达数据,结果表明ACTL6A在癌症组织中的表达水平高于正常组织(Fig 1a-c)。然后,通过对21对胃癌组织和邻近正常黏膜的分析,证实了ACTL6A在胃癌组织中的表达水平上调(Fig 1d)。细胞增殖实验表明ACTL6A的过表达对GC细胞增殖产生积极影响,敲降ACTL6A抑制GC细胞的伤口愈合和转移能力(Fig 1e-1h)。类器官实验表明,ACTL6A敲除显著降低了SNU638异种移植瘤的生长(Fig 1i-l)。

Fig 1
2. ACTL6A通过重编程GSH代谢以促进胃癌恶性进展
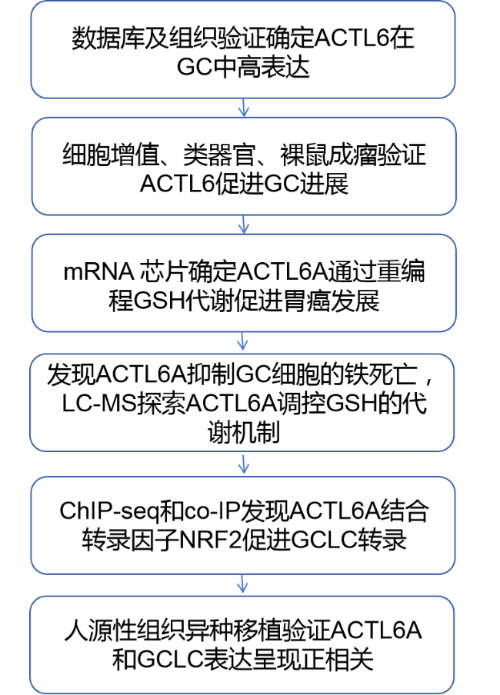
ACTL6A-KD SNU638细胞mRNA芯片结果表明ACTL6A的表达与信号通路之间的关联,其中GSH代谢是相关性最高的途径之一(Fig 2a),GC 数据集 GSE15459 和 GSE27342也有相同的结论(Fig 2b-c)。GSH通过GSH/GSSG循环保护癌细胞免受氧化应激,降低细胞内 ROS 水平并将 NADPH 转化为 NADP+(Fig 2d),并用ACTL6A-KD细胞验证了这一结果(Fig 2e-f)。DCFH-DA 结果表明,ACTL6A-KD细胞是否经过H2O2 处理都有较高的ROS 水平(Fig 2g)。此外,作者还发现N-乙酰半胱氨酸(NAC),可以重建ACTL6A-KD细胞增殖(Fig 2h-i)。NAC治疗恢复了ACTL6A-KD PDO的囊状结构(Fig 2j)。GC异种移植小鼠结果表明,ACTL6A-KD显著抑制肿瘤生长,NAC治疗又显著逆转了这种效应(Fig 2k-n)。
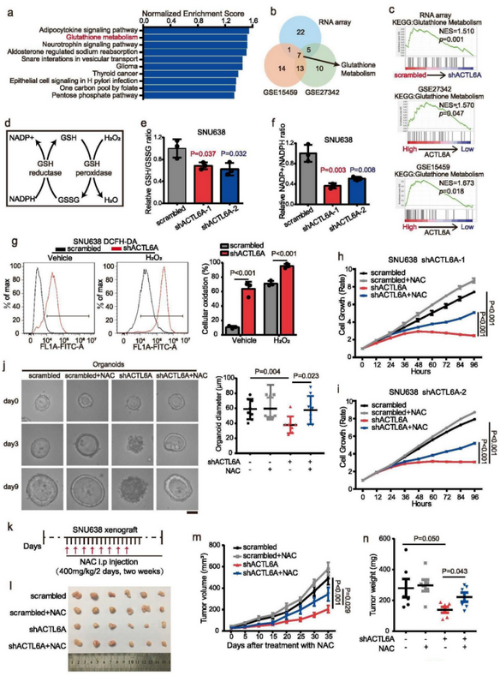
Fig 2
3. ACTL6A 抑制 GC 细胞铁死亡
为了确定 ACTL6A 是否抑制 GC 细胞的铁死亡,作者在ACTL6A消融的细胞中通过铁死亡抑制(Fer-1)处理,细胞活力显着恢复(Fig 3a),还观察到ACTL6A敲低使GC细胞对H2O2、erastin和BSO敏感(Fig 3b-f)。铁死亡的一个标志是脂质过氧化物的积累,作者对ACTL6A-KD的细胞进行了C11-BODIPY染色,发现脂质过氧化水平升高,并且在用erastin处理的细胞中这种表型更加明显,并且可以通过Fer-1处理来改变(Fig 3g),此外,Fer-1处理缓解了ACTL6A-KD诱导的PDO生长抑制,并使类器官恢复囊状结构(Fig 3h)。通过对异种移植肿瘤组织进行免疫组织化学 (IHC) 染色,发现 ACTL6A-KD会显著降低增殖标记物 Ki67 的水平,并增加增殖标记物 4-HNE的水平(Fig 3i)。
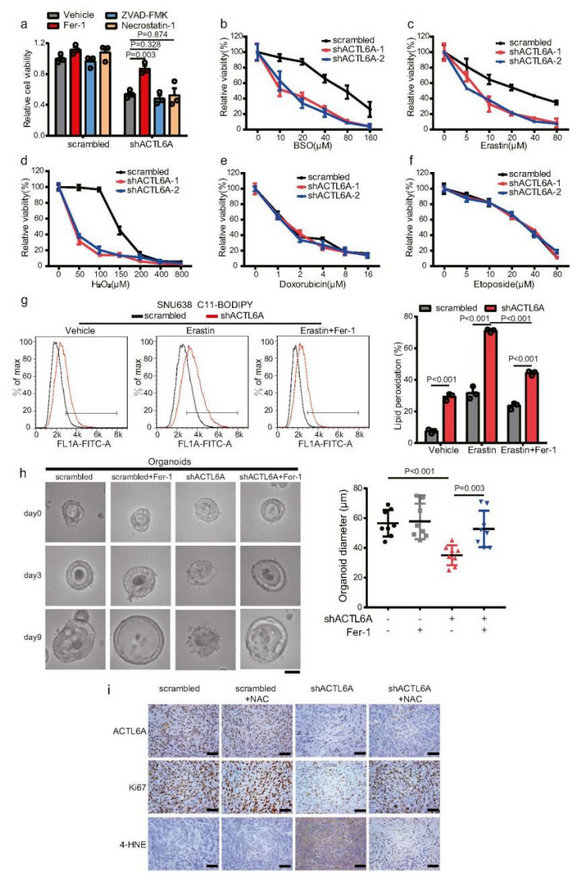
Fig 3
4. ACTL6A 通过上调 γ-谷氨酰-半胱氨酸合成来影响 GSH 从头合成
通过LC-MS评估ACTL6A调节GSH代谢的机制,研究C13-glucose的代谢去向(Fig 4a)。但是,结果未观察到C13-glucose对谷氨酸、丝氨酸或甘氨酸的总贡献发生显著变化(Fig 4b-d)。同位素实验表明,GSH的水平在表达 ACTL6A-KD的细胞中减少(Fig 4e-f)。此外,作者还研究了C13-glutamine的代谢去向,它是通过GSH从头合成产生含有5个13C原子的γ-GC和GSH(Fig 4g), C13-glutamine对谷氨酸盐总贡献未发生显著变化(Fig 4h)。同位素实验也验证了这结论(Fig 4i-j)。
Fig 4
5. ACTL6A在调控GCLC
ACTL6A-KD抑制了异种移植组织中GCLC的表达(Fig 5a),敲降后过表达GCLC,逆转了对细胞增殖的抑制(Fig 5b)。DCFH-DA染色检测,GCLC过表达恢复了敲降ACTL6A的细胞的ROS水平,并被NAC逆转(Fig 5c)。C11-BODIPY染色表明,GCLC过表达恢复了细胞的脂质过氧化水平,在Erastin处理时表型仍然显著,而Fer-1逆转(Fig 5d)。
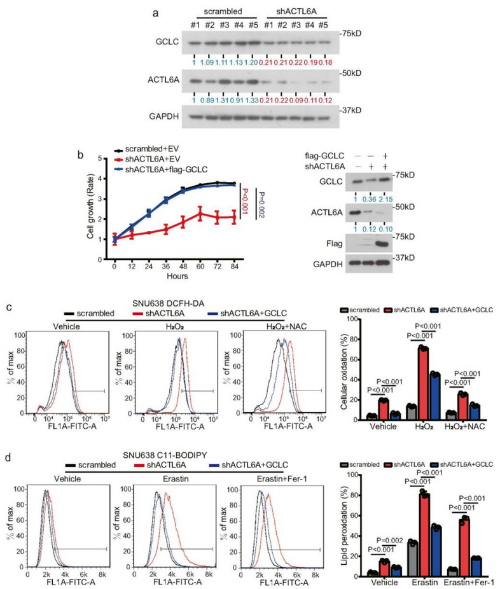
Fig 5
6. ACTL6A通过NRF2在转录水平调控GCLC
为了探索ACTL6A如何调控GCLC,作者通过对GC细胞中内源性ACTL6A进行CHIP-SEQ分析,在GCLC的转录起始点附近有一个显著的峰(Fig 6a)。Chip-seq结果表明,在GCLC的TSS附近的峰中,有一个序列与NRF2结合基序高度同源(Fig 6b),作者推测ACTL6A可能与NRF2协同作用来调节GCLC的表达。荧光报告实验验证了NRF2和GCLC的相关性。用CHIP和qRT-PCR验证,发现ACTL6A和NRF2都与GCLC TSS附近的预测结合位点相互作用(Fig 6d-e)。
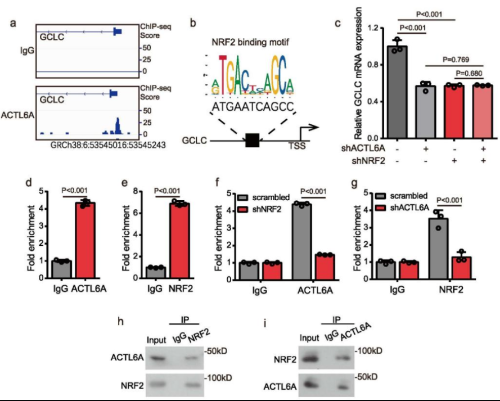
Fig6
7. ACTL6A 的 HR 结构域对于调节 GCLC 和铁死亡至关重要
构建四种缺失突变体并转染至ACTL6A-KD细胞中(Fig 7a),发现野生型(WT)ACTL6A和ΔDNBL、ΔCC和ΔNBC ACTL6A突变体逆转了敲低ACTL6A表达引起的细胞增殖抑制和GCLC蛋白表达减少,但ACTL6A HR缺失突变体没有表现出这一点恢复效果(Fig 7b-c),增加ACTL6A ΔHR缺失突变体的剂量未能逆转GCLC mRNA表达的减少(Fig 7d)。紧接着,进行了co-IP,发现ACTL6A ΔHR突变体不与NRF2结合(Fig 7e)。CHIP测定表明,与 (WT) ACTL6A 相比,ACTL6A ΔHR缺失突变体对 NRF2 与 GCLC 启动子的结合没有表现出恢复作用(Fig 7f )。之后进行DCFH-DA 和 C11-BODIPY 染色实验,发现(WT) ACTL6A 逆转了 ROS 水平和脂质过氧化水平的增加,相反,ACTL6A ΔHR缺失突变体并没有逆转这些高水平(Fig 7g-h)。
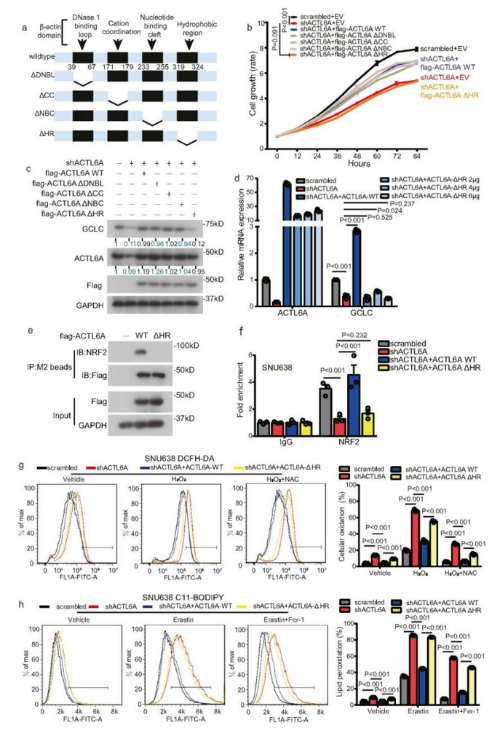
Fig 7
8. ACTL6A-GCLC-GSH 代谢轴与 GC 细胞铁死亡的临床相关性
为了确定 ACTL6A 在患者来源的PDX模型中调节 GCLC 和抑制铁死亡的临床相关性,对ACTL6A 进行免疫印记分析(Fig 8a),进行异种移植,然后注射GCLC抑制剂BSO给小鼠(Fig 8b)。BSO可以减弱肿瘤进展,相反,GCLC抑制对ACTL6A表达低的PDX肿瘤的生长影响最小(Fig 8c-f)。PDX肿瘤冰冻切片进行DCFH-DA和C11-BODIPY染色检测发现,ACTL6A高表达的PDX肿瘤的ROS水平和脂质过氧化水平较低,而BSO处理后则显着升高(Fig 8g-j)。通过LC-MS研究N15-glutamine在体内的代谢命运,它通过GSH从头合成产生含有一个N15原子的GSH肽(Fig 8k),并对高表达ACTL6A的PDX肿瘤施用BSO降低了总GSH水平(Fig 8l),N15-glutamine对GSH的总贡献减少(Fig 8m),BSO处理降低了GSH/Gln的比率(Fig 8n)。Kaplan-Meier分析表明,高 ACTL6A 和 GCLC 水平与较差的总体生存率相关(Fig 8o)。值得注意的是,GCLC 表达与 GC 中的 ACTL6A 显着正相关(Fig 8p )。
Fig 8
参考文献:
Yang Z, Zou S, Zhang Y, Zhang J, Zhang P, Xiao L, Xie Y, Meng M, Feng J, Kang L, Lee MH, Fang L. ACTL6A protects gastric cancer cells against ferroptosis through induction of glutathione synthesis. Nat Commun. 2023 Jul 13;14(1):4193. doi: 10.1038/s41467-023-39901-8. PMID: 37443154; PMCID: PMC10345109.