什么是Hi-C
染色体由DNA与组蛋白共同组成,从染色体的一级结构(绳珠模型)到四级超螺旋折叠结构,DNA分子一共被压缩了8400倍左右,正是这些折叠和压缩,导致基因在细胞中的分布复杂而又有序。
基于基因组序列,调控元件和相关的注释的信息,科学家们发现,它们在空间结构上并不是在染色体上呈线性地一字依次排开,这些离散的调控元件并不能有效地解释很多基因的调控结果和机制。由此猜测其与基因组的三维空间结构相关。
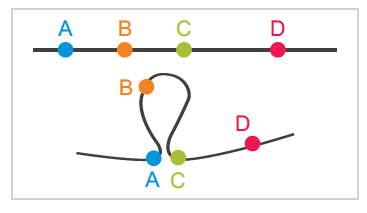
图1 loops环
基因组三维空间结构与功能的研究简称三维基因组学(Three-Dimensional Genomics, 3D Genomics)。2003年Job Dekker及其合作者提出了染色质构象捕获技术(Chromatin Conformation Capture, 3C),用于测定特定的点到点之间的染色质交互作用。随后,科学家们扩展了3C技术,开发了4C技术(Circularized Chromatin Conformation Capture),用于测定一点到多点之间的染色质交互作用。Dostie等人接着开发了5C技术(Carbon-Copy Chromatin Conformation Capture),用于测定多点到多点之间的染色质交互作用。
为了能捕获全基因组范围的染色质相互作用,Job Dekker研究组又开发出了现在大家所熟知的Hi-C技术。其是基于将线性距离远、空间结构近的 DNA 片段进行交联,并将交联的 DNA 片段富集,接着进行高通量测序,对测序数据分析可以揭示染色质的远程相互作用,从而推导出基因组的三维空间结构和可能的基因之间的调控关系。
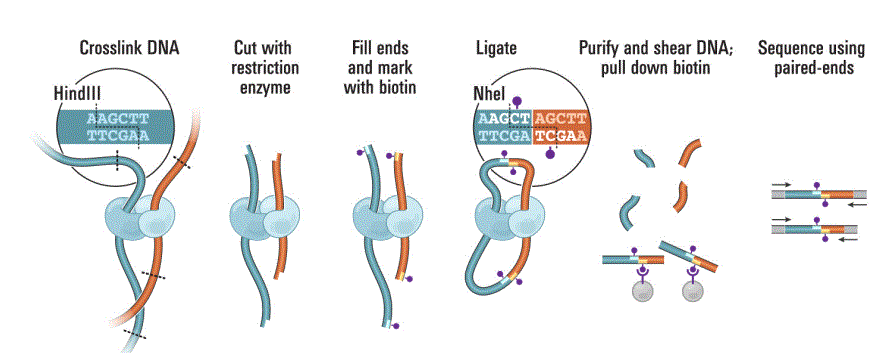
图2 Hi-C 实验流程示意图
(引自Lieberman-Aiden E, van Berkum NL, et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 2009, 326(5950):289-93)
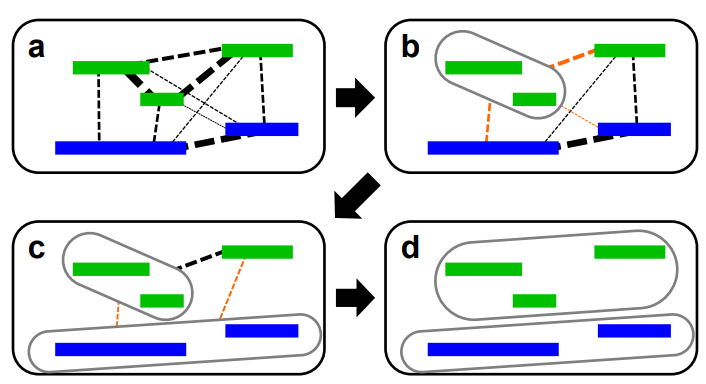
图3 序列装配过程图
(引自TingXie, Jue-FeiZheng, et al. De Novo Plant Genome Assembly Based on Chromatin Interactions: A Case Study of Arabidopsis thaliana. Molecular Plant, 2015, 489-492)
我们优化的Hi-C技术
武汉金开瑞生物工程有限公司技术顾问、华中农业大学教授, 曹罡课题组与华中农业大学李国亮教授课题组在国际著名期刊Nature Genetic(IF=27.125)上联合发表题为“Digestion-ligation-only Hi-C is an efficient and cost-effective method for chromosome conformation capture”的研究论文。该论文主要介绍了一种新的染色体构象捕获技术(DLO Hi-C),此技术信噪比高,质量控制于早期,为解析基因组三维结构提供了一种新型、高效、经济的研究方法。

图4 DLO Hi-C实验流程
新的DLO Hi-C染色体构象捕获技术(digestion-ligation-only Hi-C,DLO Hi-C),设计了巧妙的酶切位点,采用同时酶切酶连的方式,将DNA接头连接在染色体内切酶切口末端上,然后进行邻近酶连,最后再用MmeI内切酶酶切消化,回收固定大小互作DNA片段。此方法具有以下特点:1. 双交联,两次连接和消化,其中第一次同时酶切酶连,收集测序固定片段;2. 无稀释液体交联,无生物素。
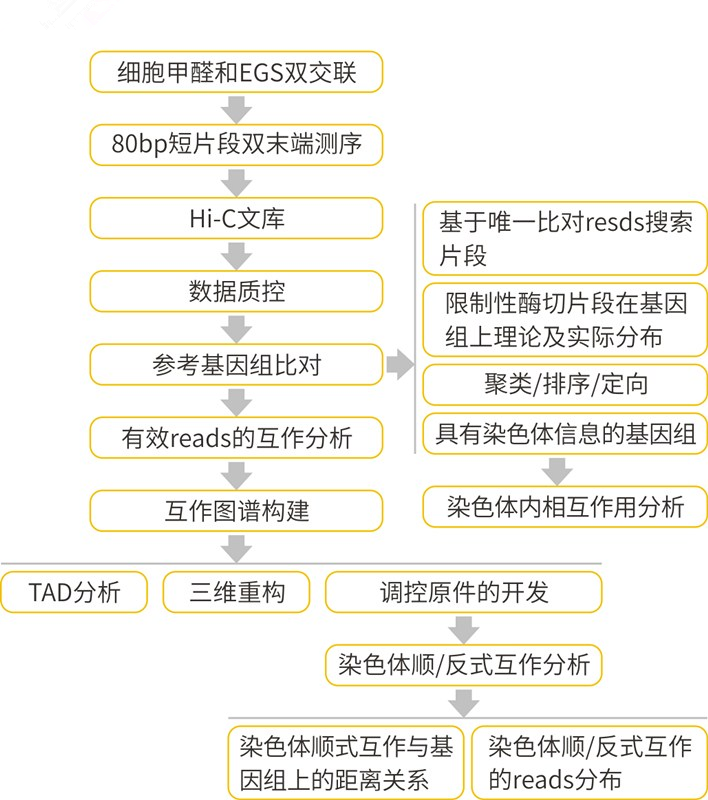
图5 Hi-C生信分析流程
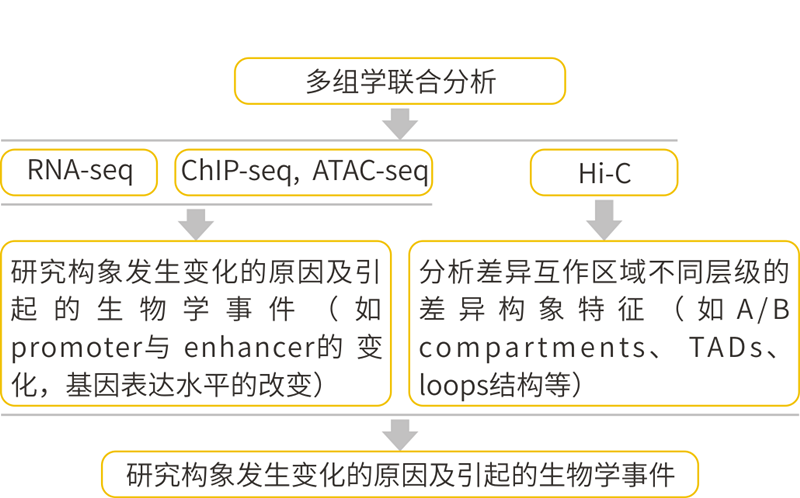
图6 Hi-C多组学分析
DLO Hi-C和其他Hi-C的衍生技术的结果比对
如表1所示,DLO Hi-C技术所能获取的图谱分辨率高于传统Hi-C技术、DNase Hi-C技术。在非冗余的有效数据读取中in situ DLO Hi-C的读取效率和in situ Hi-C相似。
表1 几种不同的Hi-C衍生方法的对比分析
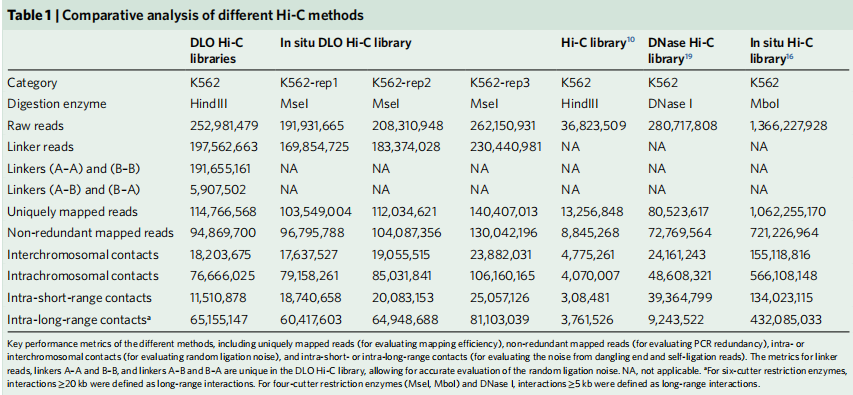
(引自Lin D, Hong P, et al. Digestion-ligation-only Hi-C is an efficient andcost-effective method for chromosome conformation capture. Nature Genetics, 2018 , 50 (5))
最终所呈现的图谱如图7所示。in situ DLO Hi-C和in situ Hi-C所呈现的矩阵清晰度相似,分辨率较高。

图7 几种不同的Hi-C的衍生方法的矩阵图
(引自Lin D, Hong P, et al. Digestion-ligation-only Hi-C is an efficient andcost-effective method for chromosome conformation capture. Nature Genetics, 2018 , 50 (5))
三种Hi-C 技术在读取A/B compartments数据时,无论原始数据量多大,所读取的数据相似,且2787个数据在三种技术中均被一致读取。
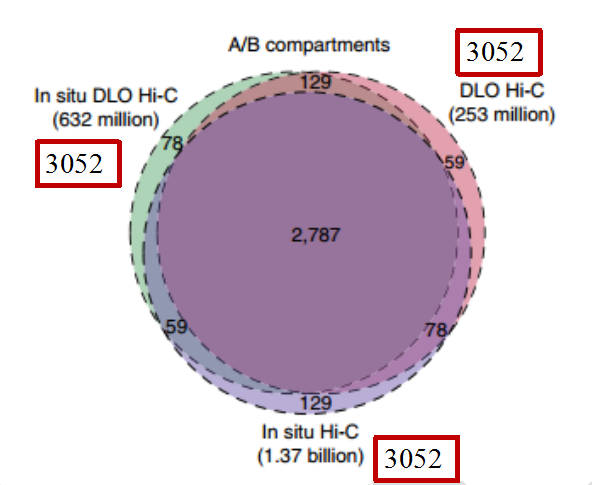
图8 三种Hi-C技术关于A/B compartments的读取
(引自Lin D, Hong P, et al. Digestion-ligation-only Hi-C is an efficient andcost-effective method for chromosome conformation capture. Nature Genetics, 2018 , 50 (5))
三种Hi-C 技术在读取TADs数据时,in situ DLO Hi-C所读取的TADs区最多是3244个。DLO Hi-C的TADs区的读取率最高,且三种技术读取了2334个一致的TADs数据。
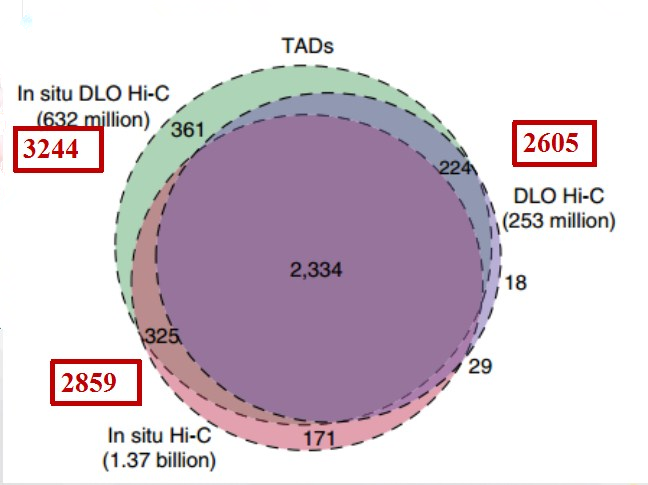
图9 三种Hi-C技术关于TADs的读取
(引自Lin D, Hong P, et al. Digestion-ligation-only Hi-C is an efficient andcost-effective method for chromosome conformation capture. Nature Genetics, 2018 , 50 (5))
三种Hi-C 技术在读取Loops数据时,in situ DLO Hi-C所读取的loops最多是12780个。这三种技术共同读取的Loops较少,929个。
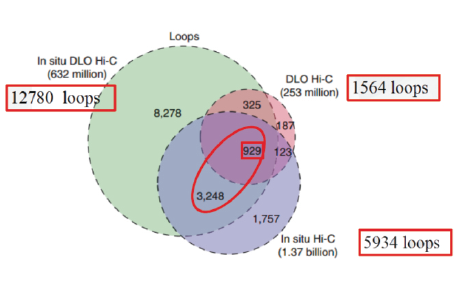
图10 三种Hi-C技术关于Loops的读取
(引自Lin D, Hong P, et al. Digestion-ligation-only Hi-C is an efficient andcost-effective method for chromosome conformation capture. Nature Genetics, 2018 , 50 (5))
综上所述,DLO Hi-C技术可获取的有效的制作矩阵的数据、A/B compartments数据、TADs数据和Loops数据较优于其他Hi-C的衍生技术。
相关阅读
如此强大的Hi-C,到底该如何运用于生物科学研究中呢?欲知详情,且听下回分解,敬请期待!