
背景
人类癌细胞系长期以来一直是癌症进展研究和药物发现的工具,但由于癌细胞系的建立涉及到对适应体外培养条件的肿瘤细胞的选择,因此通常认为癌细胞系是同质的,无法保持原肿瘤的异质性。然而,已经证实细胞系可以进化并发展成不同的细胞系。最近研究表明,通过使用单细胞RNA测序(scRNA-seq)和荧光激活细胞分选(FACS)等技术可以在已建立的细胞系中检测细胞多样性。细胞系内转录组异质性促进了上皮-间质转化和耐药的关键调控因子的发现,使这些细胞系可能成为研究药物敏感性的分子机制和测试不同治疗方案的合适模型。因此,了解常用人类癌细胞系的异质性不仅将为基于细胞系的生物医学研究提供重要信息,而且有助于确定合适的模型来研究特定癌症表型的新机制。
肿瘤内异质性的起源通常上被认为是遗传方面的,但已经扩展到表观遗传学和微环境的急剧变化。尽管scRNA-seq可以有效地解决转录程序的异质性,但它无法揭示潜在的驱动力,如表观遗传因素。为此,需要捕捉细胞间表观遗传景观变化的技术。由于染色质可及性在很大程度上可以反映转录因子结合、组蛋白修饰和DNA甲基化,并为基因调控机制提供了更深入的了解,转座酶可及性染色质单细胞测序分析(scATAC-seq)最近成为单细胞分辨率表观基因组分析中使用最广泛的分析方法。与scRNA-seq相比,scATAC-seq的一个主要优势是它提供了转录因子调控基因的机制。scRNA-seq和scATAC-seq在癌症标本中的联合应用有助于识别精确的顺式调控元件和靶基因,以及识别控制肿瘤发展的关键调控网络。
主要技术
scRNA-seq;scATAC-seq;癌细胞异质性
研究结果
1. 单细胞转录组测序揭示癌症细胞系内的潜在转录异质性
本研究选取了分布在9个谱系中以实体肿瘤为主的40个人癌细胞系和2个人正常细胞系进行scRNA-seq分析(图1a)。为了提高scRNA-seq实验的通量和降低成本,每次运行scRNA-seq时,收集来自不同谱系的三株细胞系,然后根据其表达特征计算分配给相应的细胞系(图1b)。总共获得了23,089个细胞,平均每个细胞系513个细胞,34,641个转录本,每个细胞捕获5859个基因,显示了该数据集的高质量。为了进一步检验我们的scRNA-seq实验的可重复性,在两个独立的实验中分析了三个细胞系,包括Caco2、SCC-4和MDA-MB-231。结果显示,同一细胞系的细胞在两个独立的实验运行中测量是混合的(图1c)。为了进一步了解特定的癌症谱系,分别在UMAP中绘制了乳腺癌和结直肠癌细胞系。对于乳腺癌细胞系,luminal A (LA)、luminal B (LB)、Her2+ (H)、三阴性A (TNA)、三阴性B (TNB)等相同分子亚型的细胞彼此相邻(图1d)。ESR1主要在LA和LB亚型中表达,而ERBB2在LB和H亚型中表达上调(图1e)。此外,分析了其他已知的具有临床相关性的基质和上皮标志物的表达(图1f)。
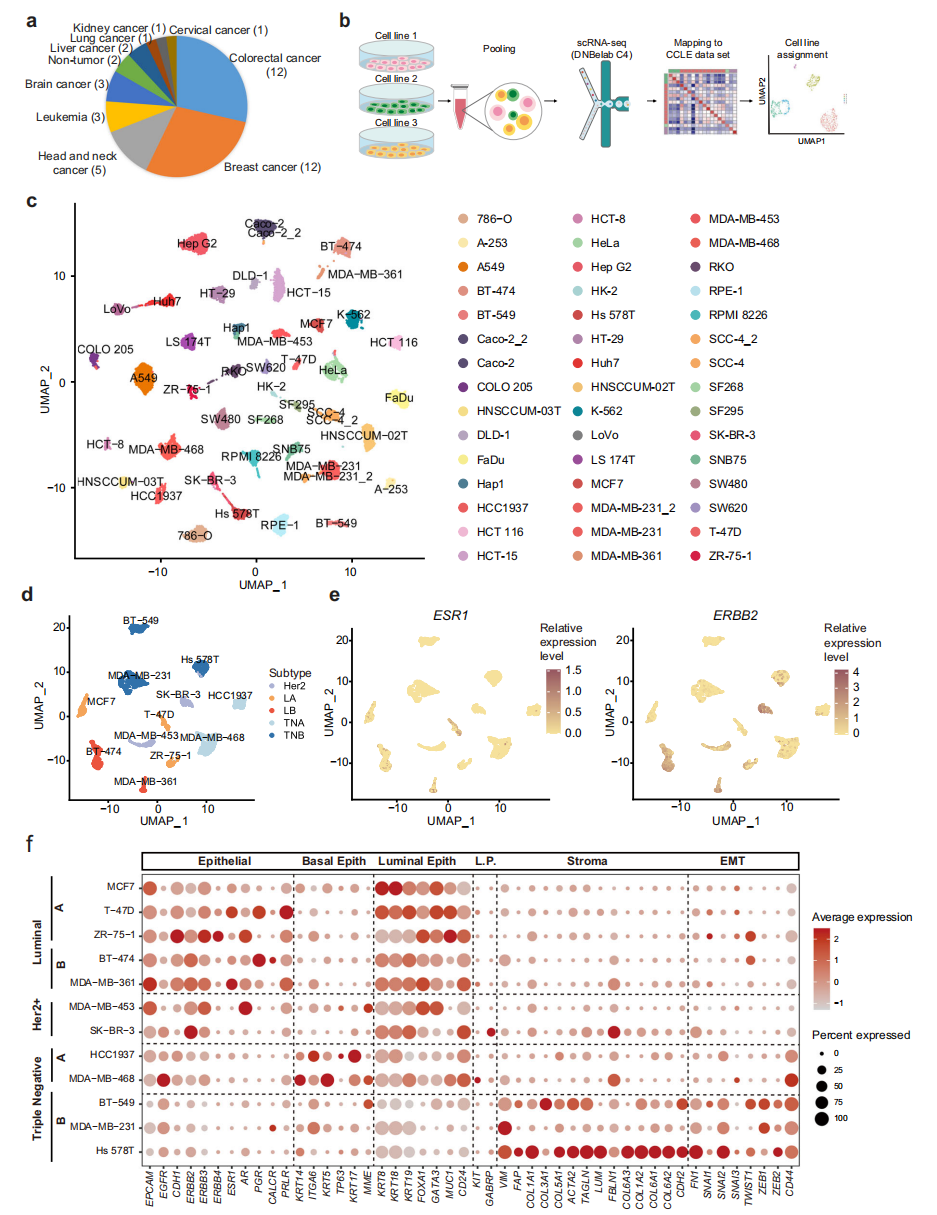
图 1
2. 单个细胞系的转录组异质性
根据转录组异质性的模式。在UMAP中,42个细胞系大致可以分为离散型和连续型两种类型(图2a)。由于这些细胞系中存在亚克隆,不同的亚簇在空间上被观察到是离散的,而连续的亚簇在亚簇之间没有明确的边界。总共有25个细胞系(57%)和17个细胞系(43%)分别属于离散组和连续组(图2b)。总体而言,离散/连续分类与多样性得分相关性较好,其中多样性得分最高的前25%的细胞系大多数是离散模式(图2b),并且离散组的多样性评分明显高于连续组,这表明具有高度异质性的细胞系通常派生出不同的亚类。然而,也有一些离散细胞系的多样性评分较低,如SW620,而连续模式细胞系的多样性评分较高,如SK-BR-3(图2b)。对于前者,簇内距离明显小于簇间距离。因此,尽管在集体距离不是很大的情况下,该细胞系的多样性得分较低,但根据分类,该细胞系呈现出离散的模式。对于后者,集体距离可以很大,而簇内距离与簇间距离没有显著差异。综上所述,两个指标显示的细胞系内异质性表明,转录组多样性可能受到不同亚克隆的存在以及细胞系内在可塑性的影响,这两个因素对细胞系内异质性的贡献在单个细胞系中有所不同。
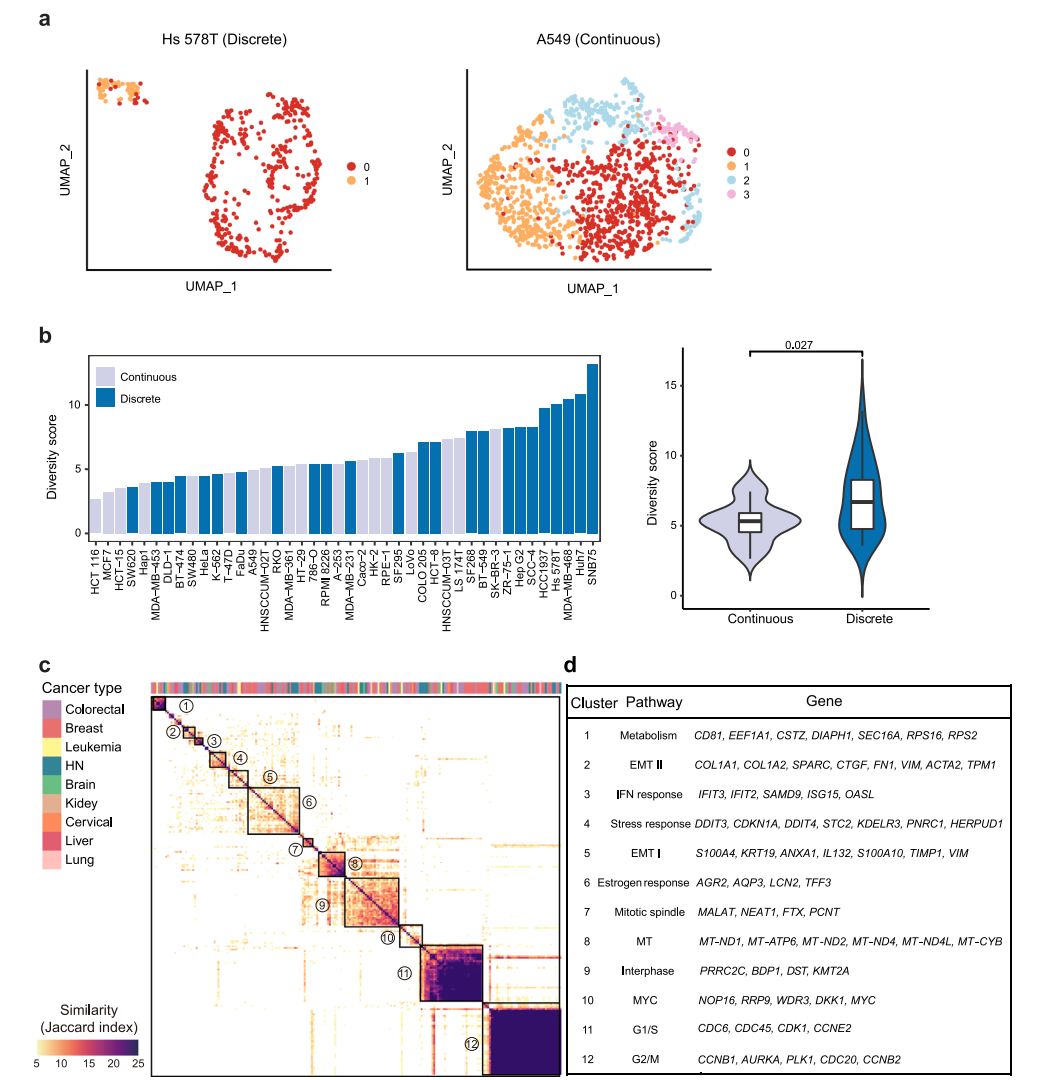
图 2
3. 转录组异质性形成的分子特征
接下来,将相似的NMF合并到NMF集群中,以确定40个癌细胞系中的复发性NMF程序。在3个以上的细胞系中鉴定出了12个异质性激活的簇(图2c)。随后,基于同一簇内单个程序之间的共有基因,其中出现在超过25%的程序中的基因被用作标志基因。大多数细胞系共有的两个最突出的簇与细胞周期有关,包括G1/S和G2/M程序(图2c、d),这表明细胞周期变化是大多数细胞系中观察到的转录组异质性的主要原因。除了细胞周期相关的程序外,另外10个簇代表了各种关键的生物过程(图2d)。
4. 人细胞系的泛癌scATAC-seq
进一步使用scATAC-seq在单细胞分辨率下研究单个细胞系内的表观基因组异质性。为了提高通量和降低scATACseq实验的成本,每次运行scATACseq时,最多合并6个具有不同表达谱的细胞系,然后根据差异表达基因的可及性特征计算分配给相应的细胞系(图3a)。经过质量过滤,获得54,597个高质量的单核,每个细胞系的中位数为1170个核(范围从203到4391个核),每个细胞核的中位数片段为11,702个。然后,我们汇总scATAC-seq谱来计算开放染色质区域的基因组分布,结果显示35.5%的ATAC峰定位在启动子-近端区域(<±1.5 kb TSS)。使用UMAP将39个细胞系一起绘制,以呈现染色质水平上的细胞系间距离,每个细胞系形成一个不同的簇(图3b)。为了在表观基因组水平上评估细胞系内的异质性,我们将细胞划分为细胞系内的初步集群。结果,62%的细胞系表现为无差别模式,38%的细胞系表现为差异模式(图3c)。总体而言,不加区分差异分类与多样性评分相关性良好,属于差异组的细胞系的多样性评分明显高于不加区分组的细胞系(图3d、e),这表明具有高度异质性的细胞系通常派生出不同的亚簇。
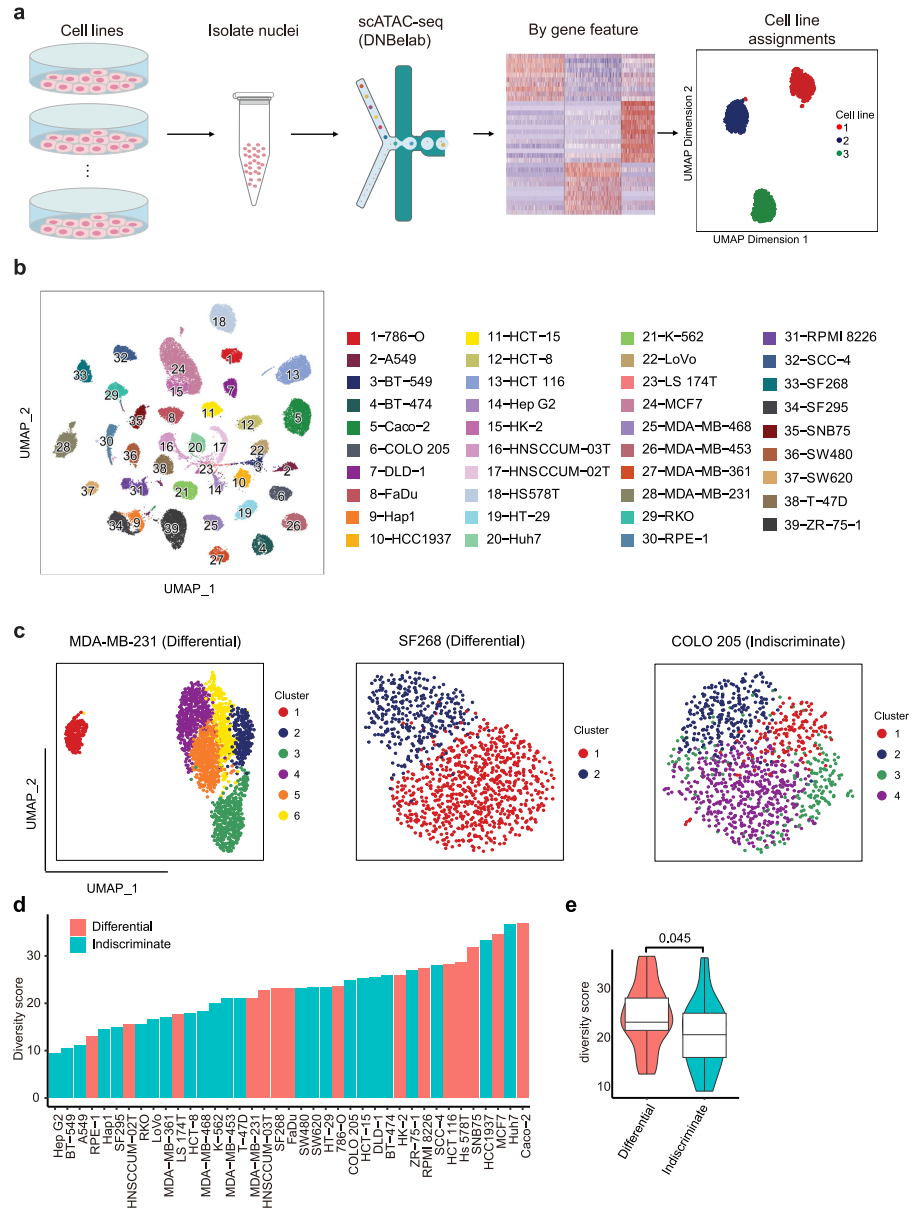
图 3
5. 与CNVs相关的转录组异质性
为明确观察到转录异质性的起源,我们首先转向恶性肿瘤常见的遗传变异。使用scRNA-seq数据分析大规模拷贝数变异(CNV),计算了与正常细胞相比,每个位点周围400个基因平均表达水平。该分析在40个(62.5%)癌细胞系中鉴定出25个CNV亚克隆(图4a、b),表明CNV诱导的拷贝数变异在很大程度上有助于转录变异;17个(42.5%)细胞系未显示相关性(图4b)。在这25个细胞系中,分别有18个和7个细胞系表现为离散型和连续型,其余15个细胞系中分别有6个和9个细胞系表现为离散型和连续型。这表明,当遗传变异导致细胞系内部异质性时,细胞系倾向于以更离散的方式呈现异质性转录组活性。然后,研究了位于CNV区域的基因是否有助于这些匹配细胞系的转录组变异性。结果显示,差异表达的基因确实在CNV区域显著富集(图4c),表明遗传变异是决定离散型细胞系细胞系内异质性的重要因素。这一事实也表明,即使对于那些具有不同模式的细胞系,也存在其他因素影响转录组异质性。
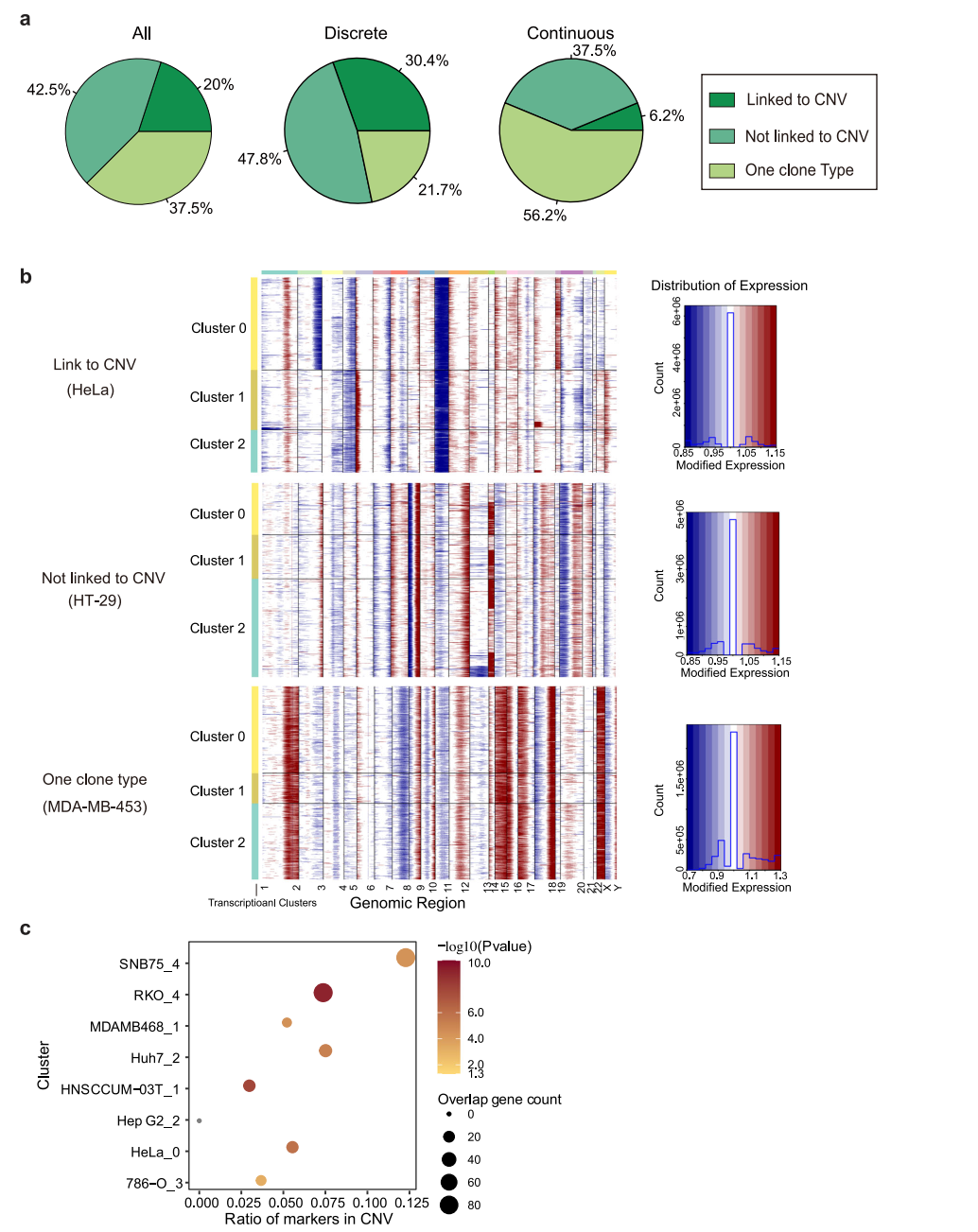
图 4
6. 染色质可及性调控转录组异质性
为了探索表观基因组异质性的机制,我们采用了包括ArchR28和ChromVAR30在内的两种方法,试图推断驱动细胞亚簇之间差异染色质可及性的潜在转录因子,并重点研究了至少三种细胞系中出现的常见TF(图5a)。参与调节IFN应答的IRF1、IRF2、IRF9和STAT2,在LS 174T和hnscum - 02t细胞系中被异质激活。然后,我们将重点放在分别在scRNA-seq和scATAC-seq数据中表现出离散和差异模式的7个细胞系上,试图在两个数据集之间匹配子簇。根据scATAC-seq数据计算基因评分矩阵,然后根据基因评分与基因表达的相似性进行比对。结果,在7个细胞系中的3个,包括MDA-MB-231、RPMI 8226和SNB75,细胞亚簇可以在两个数据集之间自如匹配(图5b、c、d)。根据scATAC-seq和scRNA-seq数据,MDA-MB-231细胞分别聚为4个和3个亚组,其中scATAC-seq数据集的亚组2与scRNA-seq数据集的亚组1和2的混合匹配。对于SNB75,,scATAC-seq数据的3亚组与scRNA-seq数据的0和1亚组的混合匹配,其余scATAC-seq数据的亚组与scRNA-seq亚组呈一对一的相关性(图5b、c、d)。这表明染色质可及性和CNV的异质性可以独立促成转录组异质性,对于这三种细胞系,染色质可及性对转录组异质性有很大贡献。
为了进一步探索驱动异质染色质可及性和RNA表达的分子机制,我们试图推断结合差异可及性染色质区域的潜在TF。通过测量基因组特征集内染色质可及性的增益或损失来推断TF活性,同时控制技术偏差,揭示潜在的TF在亚群之间表现出可变性(图5e)。在MDA-MB-231中,发现FOXA2的活性在0亚组中特异性上调(图5f),FOXA2的启动子可及性在0亚组中也有所增加(图5g)。我们随后分析了FOXA2的下游靶标,发现它们与“Kras_signaling_up”相关的基因更多相关(图5h),这些基因在0亚组中表达上调(图5i)。FOXA2可以在肿瘤发育过程中协同KrasG12D的激活。我们的结果表明FOXA2可能在控制异质性中起重要作用。
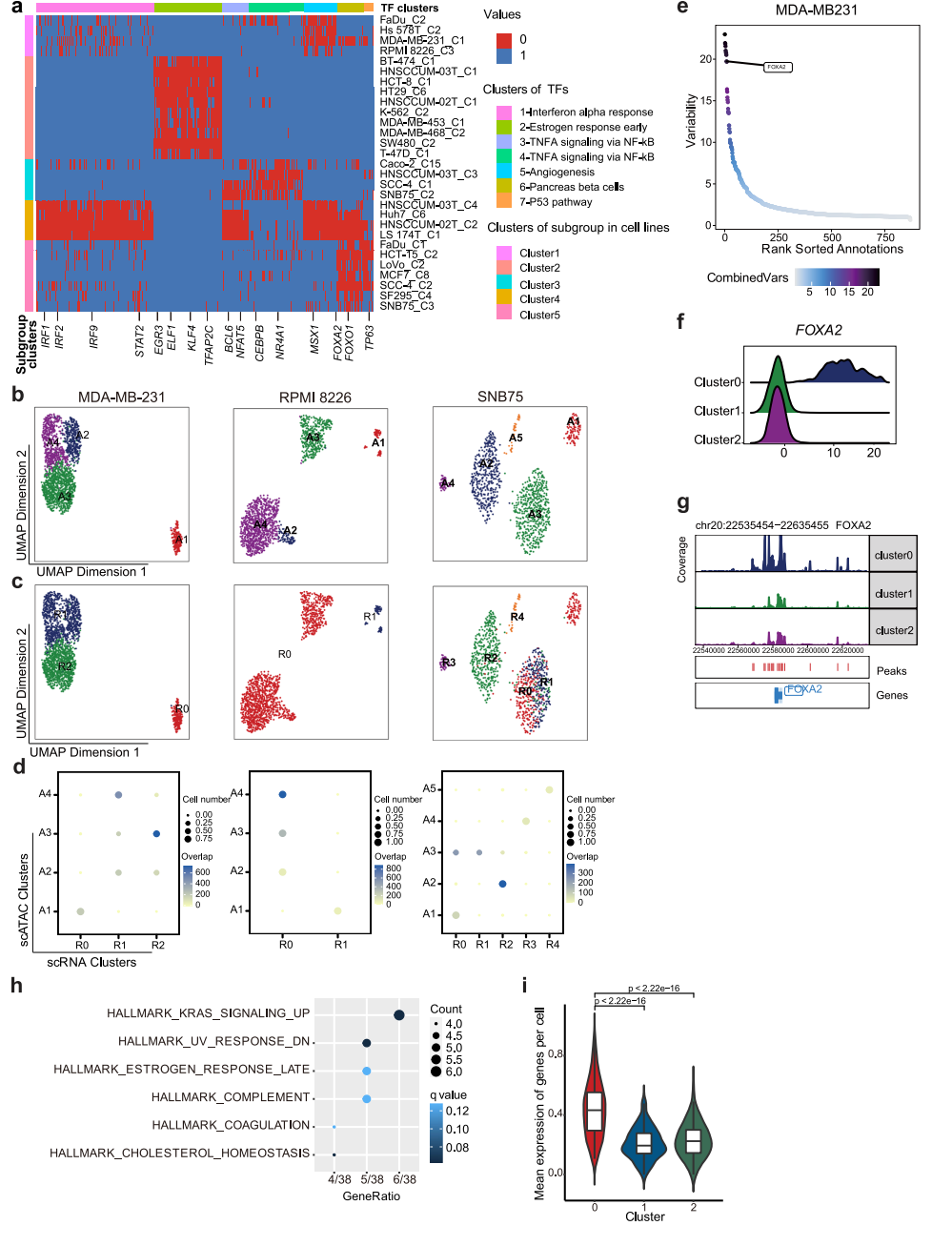
图 5
7. ecDNA分布诱导的转录组异质性
最近的研究表明,癌基因扩增既存在于染色体上,也存在于染色体外环状dna (ecDNAs)上。由于在ecDNA中没有着丝粒,与染色体扩增子相比,它们不太稳定,并且随机地分离到子细胞,这可能是细胞异质性的潜在驱动力。为了研究ecDNA对转录组异质性的贡献,我们首先建立了一种分析方法,该方法能够利用scATAC-seq数据检测高潜力的ecDNA区域,基于以下假设:(1)ecDNA高度扩增;(2) ecDNA具有更高的可及性。根据scATAC-seq数据,在每个单细胞中读取覆盖率呈现多个峰,其中第一个峰被认为是两个拷贝基因组区域的读取覆盖率,因此在这里定义为参考覆盖率。为了寻找潜在的ecDNA片段,我们首先将整个基因组分成100,000 bp的bin,在归一化测序深度后量化每个细胞中每个bin的读取覆盖率。然后,我们计算相对读取覆盖率,作为每个bin的覆盖率与单元的参考覆盖率的比率。由于ecDNA具有更高的可及性,相对覆盖率至少为6的区域被定义为潜在的ecDNA片段,并通过TSS富集评分和细胞数量进一步过滤。此外,考虑到scATAC-seq数据的稀疏性,如果由于实验和作图偏差,该区域没有被scATAC-seq读取均匀和充分覆盖,即使来自连续基因组区域的ecDNA也可以基于scATAC-seq数据检测到多个ecDNA片段。因此,我们已经鉴定的ecDNA片段的数量应该高于在细胞中发现的不同ecDNA物种的数量。
为了解决这个问题,研究了它们在单个细胞系中不同潜在ecDNA片段之间的读取覆盖率的相关性,并将附近和高度相关的片段合并为潜在的ecDNA区域。然后,我们将合并的区域定义为潜在的ecDNA区域。如图6a所示,在31个细胞系中,每个细胞系中检测到229个潜在的ecDNA区域。我们的方法成功地发现了实验证实的ecDNA,如K562中的chr9:130700000-131400000,其中ABL1和NUP214位于11中。致癌基因在潜在的ecDNA区域显著富集(图6a)。与没有致癌基因的ecDNA相比,在单个细胞系中,含有致癌基因的ecDNA出现在更高比例的细胞中(图6b),这表明含有致癌基因的ecDNA为细胞提供了生长优势,因此具有异质性。考虑到ecDNA可以通过增加DNA拷贝数来增加转录RNA的数量,我们进一步计算了ecDNA上的基因表达与ecDNA在每个细胞中的相对覆盖数之间的相关性,发现它们是部分相关的(图6c)。例如,位于SCC-4细胞系不同ecDNA中的基因KRT14和KRT17、LGALS2、SERPINE1和PCOLCE在SCC-4细胞系中以不同的三个亚簇特异性表达(图6d)。鉴定这些基因高表达的细胞比例与含有这些基因的ecDNA的细胞比例高度相关(图6e)。这一结果表明,ecDNA的高拷贝数有助于ecDNA中基因的高表达。为了进一步评估ecDNA对转录异质性的影响,我们选择了MDA-MB231,其scATAC-seq和scRNA-seq数据可以整合(图5b-d),以反映scRNA-seq数据分析的UMAP中ecDNA的拷贝数。如图6f所示,Chr12: 5900000_7200000 ecDNA在亚簇0中特别富集。同时,位于该ecDNA上的CD9、LTBR、PTMS、TPI1、ENO2、C12orf57等基因在亚簇0中也有高表达(图6g), CD9是亚簇0的顶级标记基因之一。这些结果表明,ecDNAs可以影响基因表达,从而影响细胞聚集。
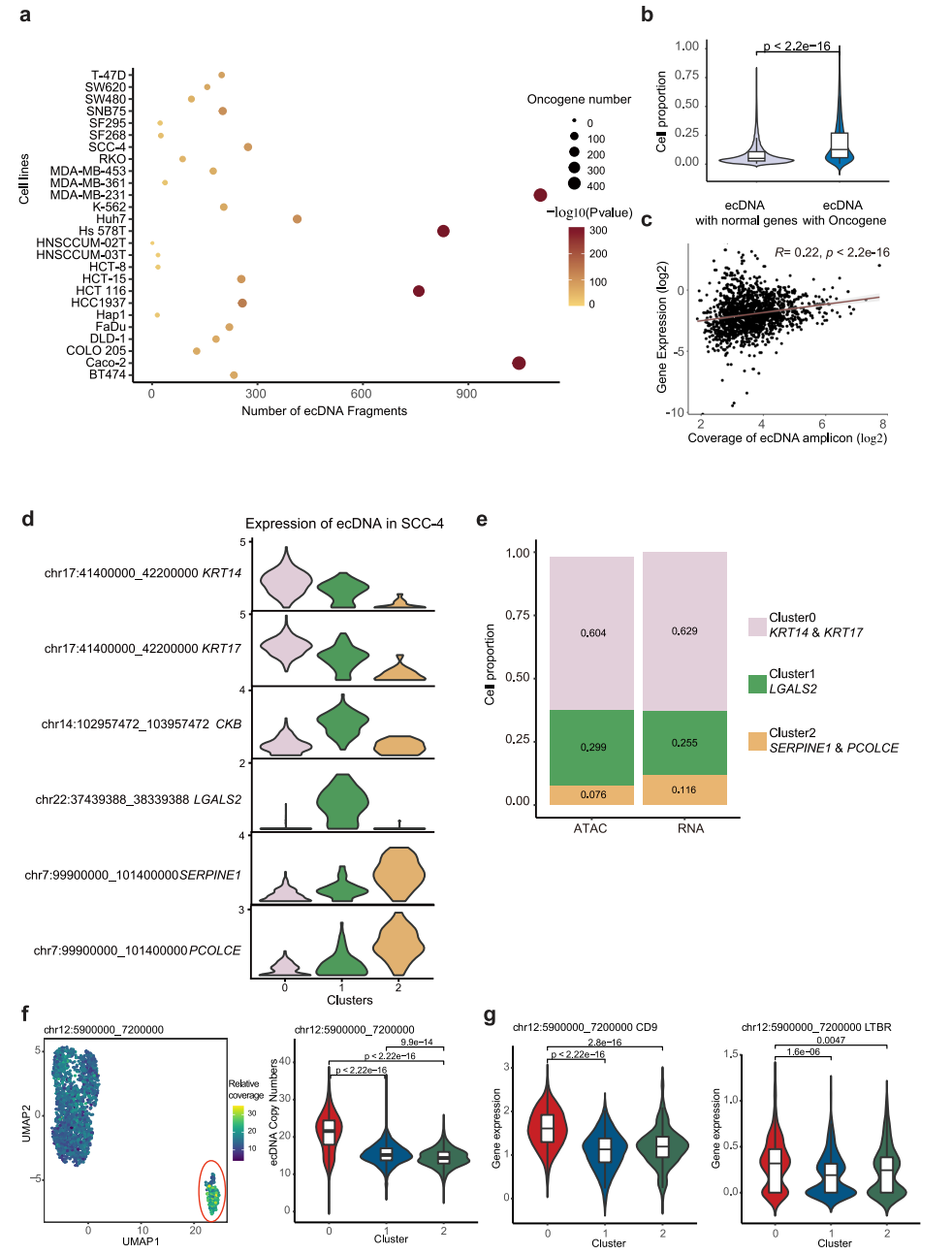
图 6
8. 低氧处理重塑细胞系内异质性
肿瘤的发生和发展是一个动态的过程,伴随着微环境的剧烈变化。微环境在肿瘤进展、治疗抵抗和转移形成中发挥重要作用,并增加了另一层调控,可驱动肿瘤内异质性。在这里,我们试图评估这些转录组异质性在不同条件下是静态的还是可塑性的。鉴于在实体瘤中,肿瘤细胞经常处于不同程度的缺氧状态,我们选择缺氧处理作为环境应激进行扰动,结果显示,缺氧处理后,低氧相关基因发生了显著变化。为了匹配缺氧治疗前后的细胞亚群,我们观察到两种类型的相关模式:(1)细胞簇在缺氧处理前后是一对一匹配的,这表明缺氧处理没有改变细胞簇之间的差异,如ZR-75-1(图7a);(2)一些细胞簇从一个分裂到多个,这表明一个细胞簇内的细胞可能对缺氧有不同的反应,如DLD-1和SW620(图7c、e)。为了确定在一个亚群中引起对缺氧应激差异反应的潜在机制,我们重点研究了在缺氧下分裂的亚群。例如,在缺氧条件下,DLD-1的子簇0分为2个簇(图7c)。我们检查了集群0和0-1/0-2之间的差异表达基因(DEGs):集群0和0-1之间变化最大的基因是缺氧相关基因,如BNIP3L、SLC2A1和ERO1A,而集群0和0-2之间的缺氧相关DEGs有限(图7d)。这表明原簇0中存在两组细胞对缺氧的反应不同,但在缺氧处理前的转录水平差异不明显。同样,在SW620细胞中,簇0被分为簇0-1和簇0-2(图7e);尽管所有细胞都对缺氧有反应,但簇0-1中的细胞显示了与EMT相关的其他基因的变化(图7f)。
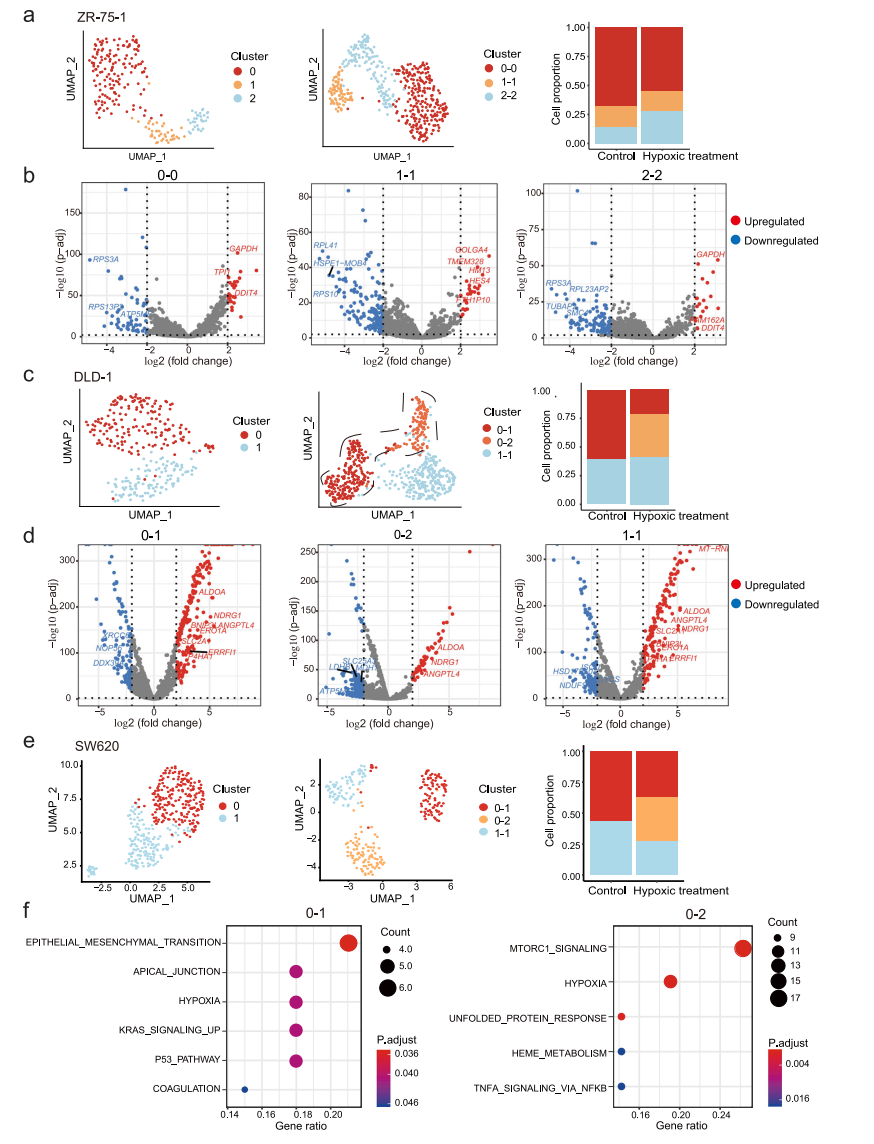
图 7
先前的研究表明,组蛋白修饰和特异性TF结合的变化可能先于基因表达的变化,并预示着基因表达的变化,这表明引发的染色质状态通过增强激活或抑制基因来影响细胞命运。在这里,通过交叉检查scRNA-seq和scATAC-seq数据,我们旨在确定这些引物可接近的染色质,这可能是对缺氧治疗的不同反应的基础。为此,我们比较了常氧条件下的scATAC-seq数据和缺氧条件下的scRNA-seq数据,并鉴定出两个数据集之间可以自由匹配的细胞亚簇(图8a、c)。在DLD-1细胞中,亚簇0-1对缺氧敏感。scATAC-seq数据显示,ETS (ELK4、FLI1)和E2F (E2F3、E2F6)家族在0-1亚簇细胞中被激活,这些细胞对缺氧敏感。这与先前的研究一致,表明ETS转录因子在缺氧诱导的基因表达中的作用(图8b)。在SW620细胞中,除了缺氧相关基因外,亚簇0-1也出现了EMT相关基因的变化。基于scATAC-seq数据,我们发现HMG (TCF4、TCF3)家族成员是集群0-1中的差异TF(图8d)。鉴于先前的研究表明HMG家族可以调节EMT,这意味着这些TF可能参与了缺氧诱导的EMT激活。这些结果表明,异质染色质可及性可能有助于表面上同质细胞群体的异质应激反应。
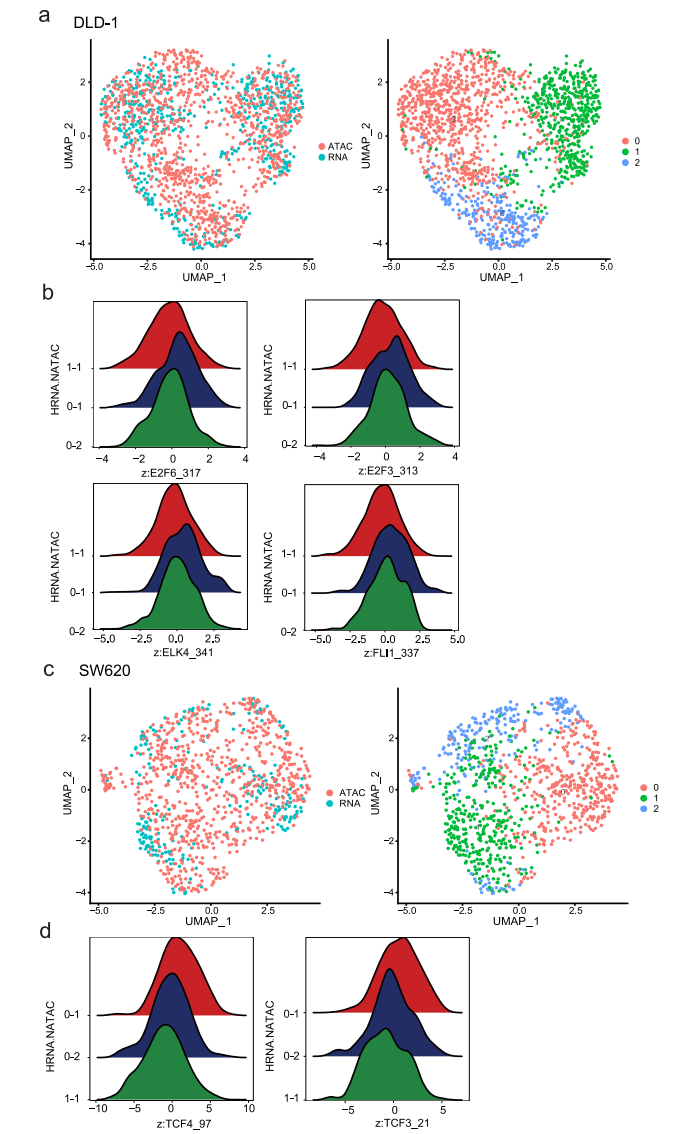
图 8
结论
为了表征不同细胞系的转录组学和表观遗传异质性,我们对数十种人类细胞系进行了scRNA-seq和scATACseq,主要包括乳腺和结直肠细胞系。通过整合scRNA-seq和scATAC-seq数据,我们研究了驱动异质性的分子机制,并发现拷贝数变异CNV仅对观察到的转录组异质性有部分贡献。表观遗传多样性和染色体外环状DNA (ecDNA)分布对细胞系内异质性有重要影响。此外,通过谱系追踪和缺氧处理,我们证明转录组异质性是可塑的,可以在环境胁迫下重塑。综上所述,我们的研究对常用的人类癌细胞系进行了单细胞多组学研究,并提供了细胞系内异质性的机制见解。
参考文献:
Zhu Q, Zhao X, Zhang Y, Li Y, Liu S, Han J, Sun Z, Wang C, Deng D, Wang S, Tang Y, Huang Y, Jiang S, Tian C, Chen X, Yuan Y, Li Z, Yang T, Lai T, Liu Y, Yang W, Zou X, Zhang M, Cui H, Liu C, Jin X, Hu Y, Chen A, Xu X, Li G, Hou Y, Liu L, Liu S, Fang L, Chen W, Wu L. Single cell multi-omics reveal intra-cell-line heterogeneity across human cancer cell lines. Nat Commun. 2023 Dec 9;14(1):8170. doi: 10.1038/s41467-023-43991-9. PMID: 38071219; PMCID: PMC10710513.