2021年11月澳大利亚Victor Chang心脏研究所Richard P. Harvey团队与Vaibhao Janbandhu等人合作在Cell Stem Cell上发表题目为“Hif-1a suppresses ROS-induced proliferation of cardiac fibroblasts following myocardial infarction”的心血管文章,该研究发现HIF-1a能够抑制心肌梗死后ROS增加介导的心脏成纤维细胞增殖所致心肌纤维化扩张的现象。该发现为心血管疾病的治疗提供了新的药物靶点和研究方向。
缺氧诱导因子1(HIF-1)通路一直在心血管生物学领域中发挥重要作用。妊娠期生理性缺氧激活局部HIF-1通路,对心脏的腔室和室间隔形态发育至关重要。成人心脏经历周期性的缺氧,包括生理上在高海拔和运动期间以及病理上在缺血期间、心肌细胞肥大、炎症和纤维化期间的缺氧,这些情况可能与ROS水平升高和氧化损伤有关。
机体通过HIF-1通路适应缺氧微环境,HIF-1α属于bHLH-PAS (basic-helix-loop-helix-Per-Arnt-Sim) 家族的转录因子。缺氧条件下HIF-1a亚基稳定并易位到细胞核,异二聚后激活下游基因通路,减少氧消耗和活性氧(ROS)生成,并恢复氧递送。
HIF-1在心脏中的作用的研究主要集中在心肌细胞上,且其在ECM重构和缺血预适应以及动脉粥样硬化进程中的作用已被证实。但HIF-1在心脏成纤维细胞中的功能尚未被阐明。心脏成纤维细胞是一种异质性的基质细胞群,通过控制细胞外基质的沉积和周转来促进心脏的生物力学完整性,并介导心脏修复和再生中的适应性纤维化。心脏成纤维细胞几乎与所有形式的心血管病理表型相关如:ECM分泌与收缩、纤维化与疤痕形成、心力衰竭等。
亮点要素
研究思路
-
通过对未损伤的小鼠S+P+阳性的CFs采用流式等分析,发现该种类细胞在缺氧状态下的代谢更倾向于糖酵解途径而非常规的线粒体氧化磷酸化途径,由此发现CFs不同于CMs的代谢谱差异。(现象挖掘)
-
HIF-1a在缺氧状态下CFs中高表达,其靶基因能在正常的氧化磷酸化中促进糖酵解途径,通过scRNA-seq结果与HIF-1靶基因数据库的比对确定HIF-1的靶基因是同样在缺氧CFs中高表达的MEIS1。(锁定目标)
-
体外利用与CFs类似的cCFU-Fs成纤维细胞系,验证线粒体质量低的细胞具有和CFs类似的增殖能力;体内构建CFs特异性Hif-1a敲除小鼠验证其在CFs稳态和心脏疾病中的作用。(体内外验证)
-
通过整理并利用qRT-PCR逐一验证与HIF-1相关的机制如糖酵解、血管生成、氧化还原稳态等,最终将HIF-1作用机制确定为调节线粒体氧化应激。(关联机制验证)
-
通过ROS探针等验证在缺氧CFs中HIF-1α调节氧化应激降低线粒体ROS,进而抑制由ROS介导PI3K/AKT/mTOR及MAPK、ERK1/2等通路激活导致的CFs增殖。(机制串联)
研究内容
首先分离正常小鼠心脏中 SCA1+/PDGFRa+/CD31-的CFs,在低氧状态下通过Pim染色流式分析发现S+P+细胞缺氧程度高于其他非心肌细胞群,其氧消耗及ATP水平远低于其他非心肌细胞群。但其葡萄糖消耗速率及细胞内乳酸水平却更高,提示S+P+细胞的代谢更倾向于糖酵解途径而非常规的线粒体氧化磷酸化途径,与之相对应的S+P+细胞的线粒体质量同样偏低。HIF-1a及其靶基因在S+P+细胞中高表达,已知HIF-1a的靶基因能在氧化磷酸化中促进糖酵解,随后通过已发表的scRNA-seq结果与HIF-1靶基因数据库的比对确定HIF-1a的靶基因MEIS1。又进一步对线粒体质量差异的细胞进行增殖能力的对比,发现低线粒体质量的细胞增殖能力更强,这也与缺氧状态下CFs的增殖现象相符合。
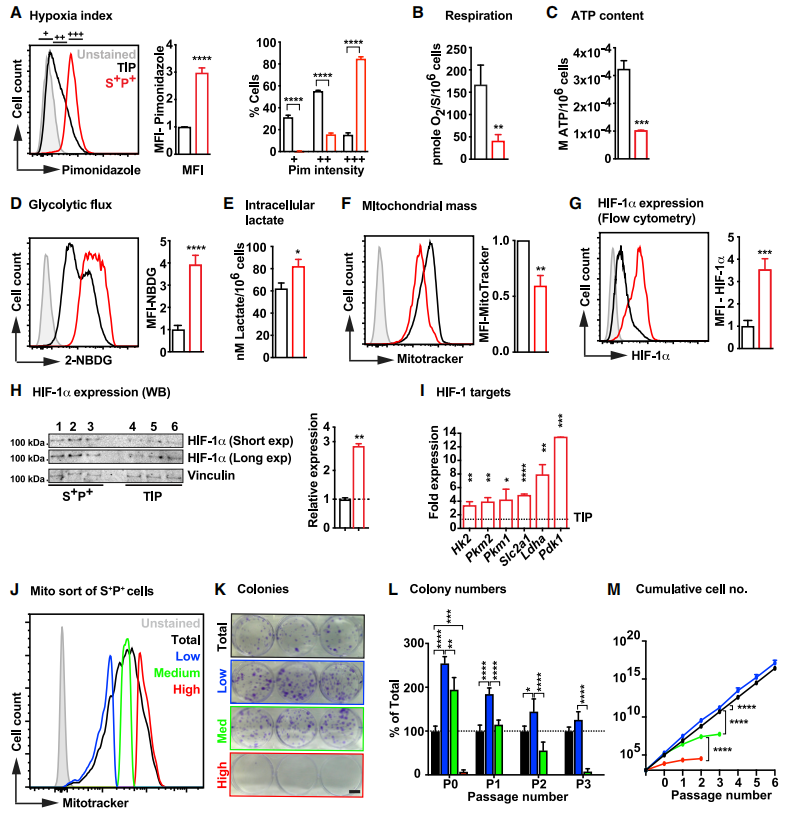
图1.成年小鼠心脏中S+P+细胞的代谢谱
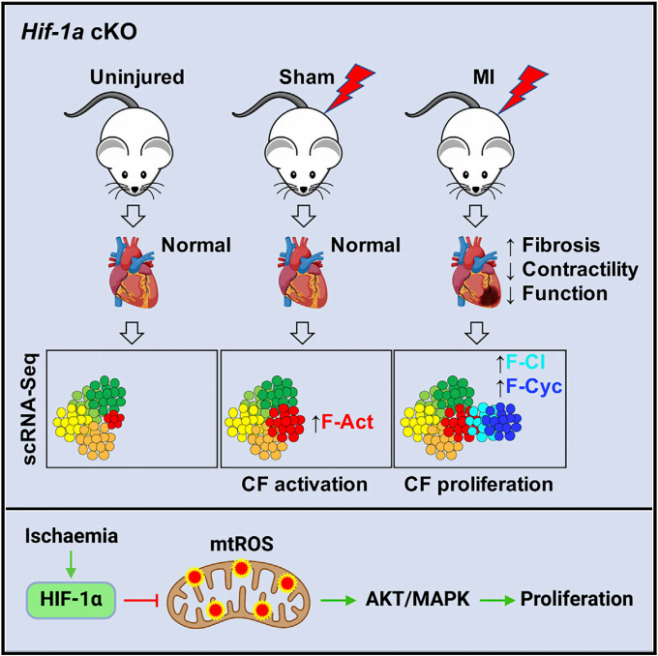
为了探索HIF-1a在心脏成纤维细胞稳态和疾病中的作用,研究者构建了CF特异性HIF-1a他莫昔芬诱导的条件性敲除小鼠并对其心脏中CF进行测序。发现cKO小鼠下调的差异基因与发表的HIF-1靶基因存在交集,且GOterm分析显示,HIF-1相关靶点参与了增殖等细胞功能的调控,其他少数上调的基因与炎症、蛋白水解和应激等途径有关。接下来为了研究cKO小鼠在心脏损伤后CFs的增殖情况,研究者构建了心肌梗死模型并利用EdU追踪增殖细胞。MI手术3天时在梗死区观察到EdU+细胞,7天时延伸至梗死边界区,而谱系阴性小鼠无变化。提示CFs中Hif-1a的缺失可导致心肌梗死后成纤维细胞增殖增加。
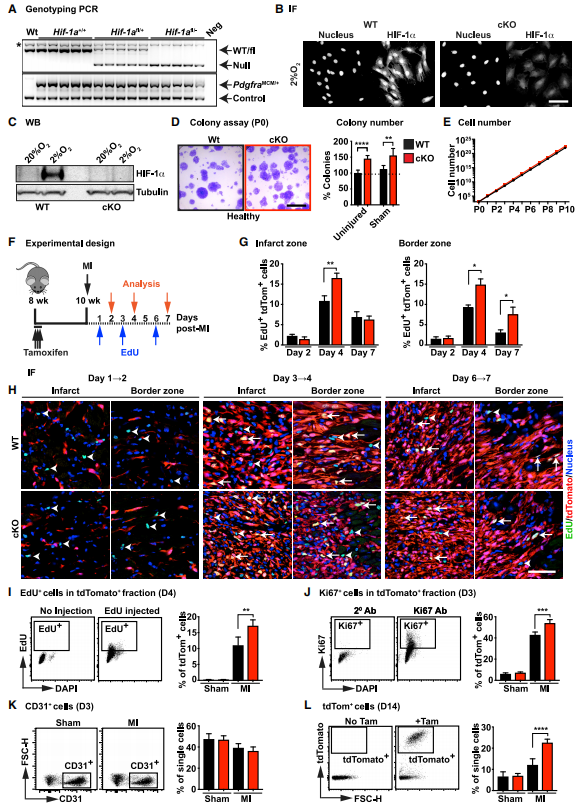
图2.心肌梗死后hif-1a缺失的CFs的增殖能力有缺陷
对MI及假手术第3天的cKO小鼠心脏成纤维细胞进行scRNA-seq测序,发现假手术组cKO小鼠CFs同样存在激活现象,验证了CFs存在全身因素的敏感性。测序的另一发现是成纤维细胞前体减少,代表损伤的中间体和激活体增加,证明更多的CF进入细胞周期。同时由于中间体和激活体增多,相关的蛋白合成基因上调,编码细胞信号传导负调控因子的基因下调,导致CF开始增殖。对心肌梗死后第14天的cKO小鼠左心室进行qPCR,发现细胞外基质、重构蛋白酶和纤维化介质相关基因显著上调,证明心脏纤维化反应增加。
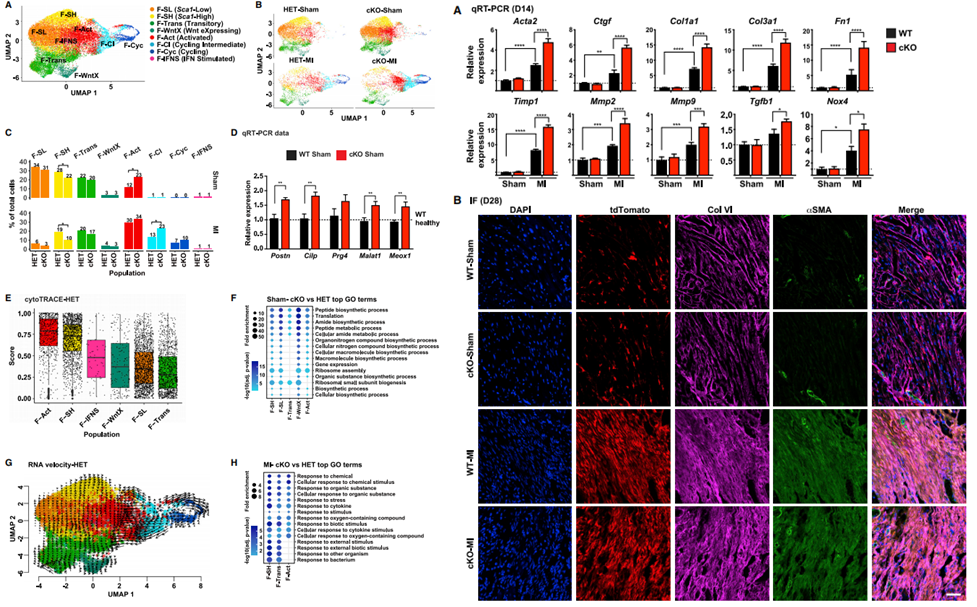
图3.HIF-1a缺失的CFs在心梗后重入细胞周期并加重心梗后纤维化
为了更直观的展现cKO小鼠CFs的动态,研究者运用前沿3D技术构建了3D工程化心肌微组织。体外培养显示cKO小鼠CFs显著扩张并伴随着胶原沉积,但并不影响心肌组织的面积。随后又探究了3D心脏体系CFs动作电位的传导情况,与WT相比cKO心脏束的传导速率较慢、动作电位持续时间更长,并表现出收缩力的损害。cKO小鼠CFs在这种缺乏血管、免疫和其他心肌细胞类型的简单3D体系中的影响与体内研究基本一致。
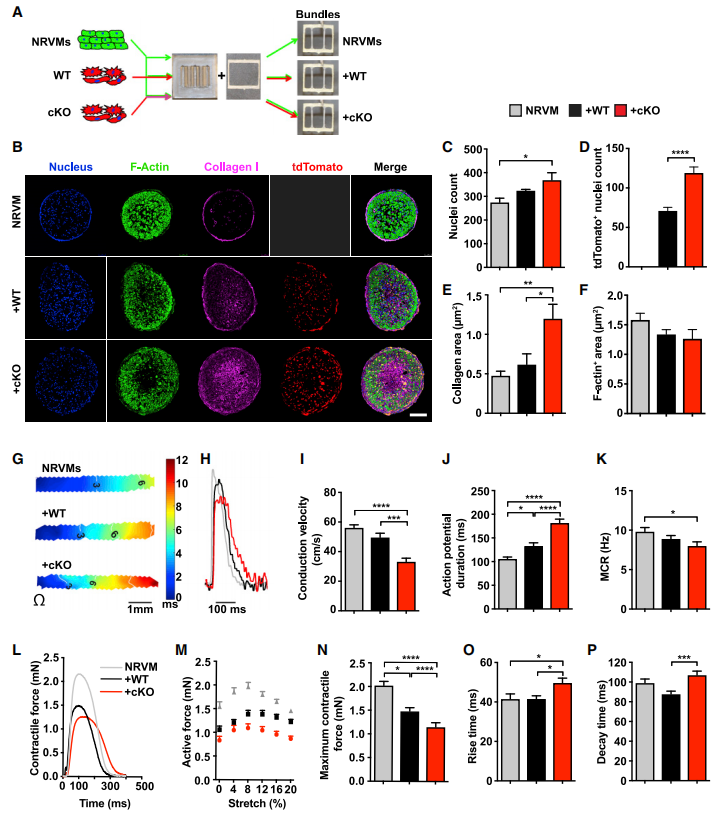
图4.Hif-1a缺失的CFs在体外显著恶化了3D心肌束的电位传导和机械力
机制研究,在应对缺氧时,Hif-1a通过确保有效利用现有的氧气和能量底物,同时最小化线粒体氧化应激来调节代谢。研究者对MI术后第1天纯化的cKO小鼠CF进行qRT-PCR检测,发现与糖酵解、血管生成和氧化还原稳态有关的HIF-1靶基因的表达显著降低。鉴于心肌梗死后cKO心脏中控制氧化还原稳态的HIF-1a靶点下调有助于减少线粒体ROS,并且已知正常生理产生的ROS可以作为干细胞/祖细胞增殖和分化的信号。研究者大胆假设ROS的增加是cKO小鼠CFs增殖和纤维化增加的原因并进行验证,线粒体特异性ROS探针证明梗死后的ROS增加定位于线粒体中并伴随CF的显著增殖。接下来利用线粒体氧化剂MitoPQ(ROS促进剂)在体处理后CF显著增殖,而线粒体抗氧化剂MitoT(ROS抑制剂)治疗后CF增殖及总数均减少。已有研究表明,ROS与PI3K/AKT/mTOR、MAPK、ERK1/2通路的激活有关。MitoT处理后,心梗cKO小鼠的CF中pERK1/2和pAKT降低到与心梗WT小鼠相同的水平。这些数据证明了ROS与AKT和ERK1/2信号通路的关系及其在CF增殖中作用机制。
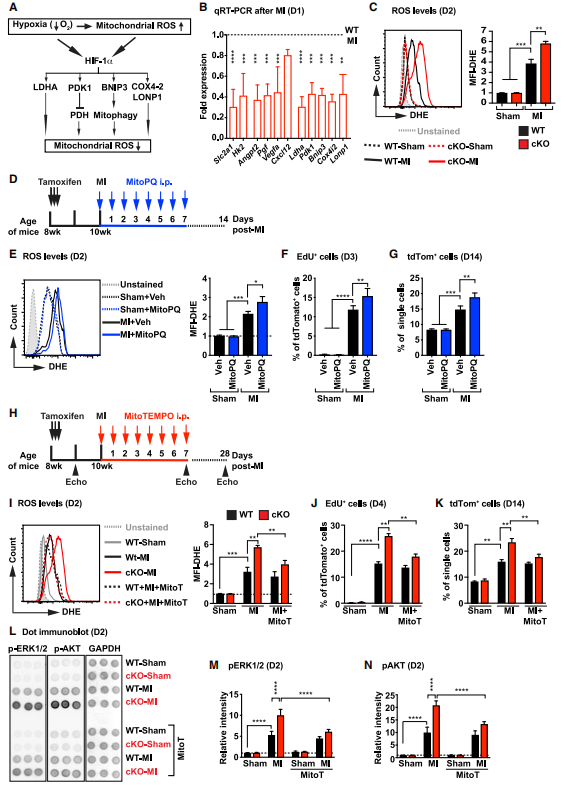
图5.Hif-1a介导的氧化还原调节控制心肌梗死后CFs的增殖
通过研究线粒体抗氧化剂MitoT为未来心脏病治疗提供解决思路。MI术后28天的小鼠心脏瘢痕面积增加、活心肌体积减少。MitoT的治疗挽救了心梗cKO小鼠的心脏心室重构、CF增殖及ECM扩张。心脏超声显示MI术后第7天和第28天,cKO小鼠心脏功能明显恶化,包括面积变化减少、左室射血分数和心输出量减少,左室收缩末期和舒张量增加。cKO-MI小鼠的心重体重比也显著升高,体现出CF增殖及ECM的扩张。MitoT的治疗能够逆转不良的心功能参数和HW/BW比值并使其恢复到WT小鼠MI术后心脏相似的水平,该发现或可为心脏病治疗研究提供新的思路。
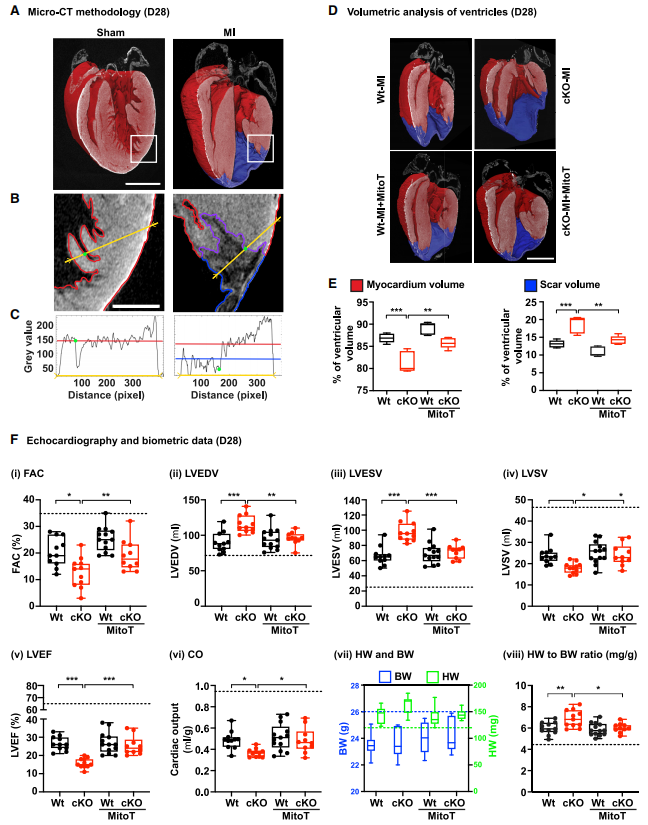
图6.MitoT治疗挽救了心肌梗死后cKO小鼠的心脏结构和功能缺陷
和元生物具有多年丰富的动物实验和心血管动物模型构建的实操经验,并兼具出色的基因操作与病毒构建等技术。同时依托公司的检测平台、病理平台、细胞平台等专业实验平台,可为您提供从心血管动物模型到基因干预、病理检测、细胞功能学研究等一站式心血管研究服务体系,您可体验文中涉及到的心血管前沿研究手段与支持。欢迎扫描下方二维码了解更多实验服务详情。